R包 VarExp
R包VarExp的实现(针对linux系统)1先把RCurl下载下载第一个圈或者第二个圈(忘了下载的那个了)2下载VarExp下载这个圈住的格式3在Rstudi
R包VarExp的实现(针对linux系统)
1先把RCurl下载
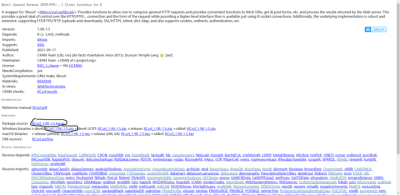
下载第一个圈或者第二个圈(忘了下载的那个了)
2下载VarExp
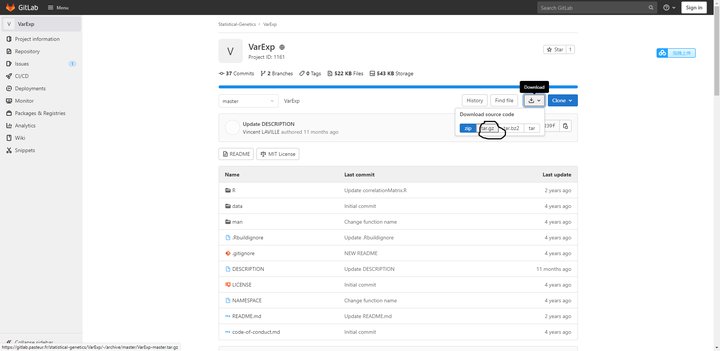
下载这个圈住的格式
3在Rstudio里安装RCurl 和VarExp
以Install去本地载入刚下载的包
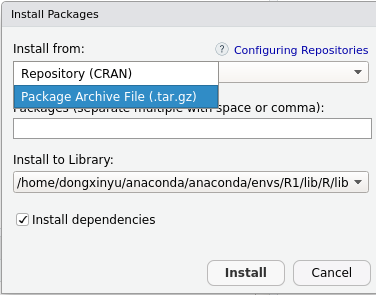
点下边这个Package Archive File
4遇到没有bitops这个程辑包错误时,在Rstudio里Install 这个包
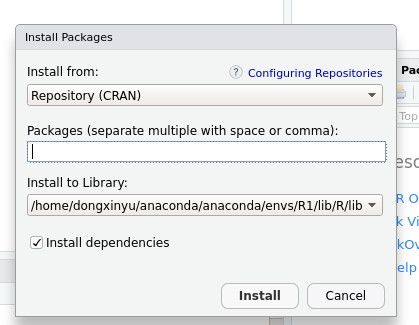
这个包就是点CRAN
5
然后继续运行会出现tabix的错误https://gitlab.pasteur.fr/statistical-genetics/VarExp/-/issues/2
这个网址里的错误跟我们一样,我们需要先下载需要下载的东西
即后边这两个网址都把东西下载了
http://bochet.gcc.biostat.washington.edu/beagle/1000_Genomes_phase3_v5a/b37.vcf/chr1.1kg.phase3.v5a.vcf.gz
http://bochet.gcc.biostat.washington.edu/beagle/1000_Genomes_phase3_v5a/b37.vcf/chr1.1kg.phase3.v5a.vcf.gz.tbi
然后下载完后还需要需要运用samtools软件里的tabix(这个软件是linux系统下的)
所以下载这个软件,往linux里输入
conda config --add channels bioconda
conda config --add channels conda-forge
conda install samtools==1.11
(上边这里的三条命令输入到你linux系统里,然后成功后就有samtools了。这命令的基础是我安装了conda的环境)
接下来的是在R语言输入的东西
old_path <- Sys.getenv("PATH")
Sys.setenv(PATH = paste(old_path, "这里是你安装的samtools的所在位置", sep = ":"))
C <- getGenoCorMatrix(GWAS$RSID, GWAS$CHR, GWAS$POS, GWAS$A0, "EUR", pruning = FALSE, web=FALSE, path='~/Downloads/chr1.1kg.phase3.v5a.vcf.gz')
这里最后一行的path指的是你下载的chr1.1kg.phase3.v5a.vcf.gz的所在路径
然后就可以一直运行了




